


Abstract and Introduction
Abstract
Purpose: This study evaluated safety, pharmacokinetics, and efficacy of 2 dose schedules and 2 infusion times of panitumumab in patients with advanced solid malignancies.
Patients and Methods: This phase I multicenter, openlabel study sequentially enrolled patients with advanced solid tumors refractory to standard therapy, or for which no standard therapy exists, to receive panitumumab 6 mg/kg every 2 weeks or 9 mg/kg every 3 weeks. Patients receiving panitumumab every 2 weeks received either all infusions over 60 minutes or a 60-minute infusion for the first dose followed by 30-minute infusions if the first infusion was well tolerated. Patients in the every-3-week cohort received 60-minute infusions. Safety outcomes included the incidence of adverse events and antipanitumumab antibody formation. Pharmacokinetic properties were determined. Efficacy endpoints included response rate and duration of response.
Results: Eighty-six patients were enrolled; 84 (98%) received panitumumab. Treatment-related adverse events occurred in 90% of patients. Safety profiles were similar between patients receiving 30-minute (n = 20) and 60-minute (n = 43) infusions every 2 weeks and patients receiving panitumumab every 3 weeks (n = 21). Panitumumab exposure at steady state increased dose proportionally, and peak serum concentrations were similar in patients receiving either 30- or 60-minute infusions every 2 weeks. Objective responses were seen in 4 patients (5%) with colon, rectal, esophageal, and bladder cancers.
Conclusion: Similar drug exposures and safety profiles were observed in patients receiving panitumumab 6 mg/kg every 2 weeks with either 30- or 60-minute infusions and antitumor activity was seen in some patients. Exposure increased approximately dose proportionally at steady state.
Introduction
The human epidermal growth factor receptor (EGFR) is a member of a subfamily of type-1 receptor tyrosine kinases, which includes EGFR (ErbB1), HER2/neu (ErbB2), HER3 (ErbB3), and HER4 (ErbB4).[1] Once bound by ligand, EGFR is autophosphorylated and initiates a signal transduction cascade that promotes cell proliferation and survival. Overexpression of EGFR has been found to occur in many tumor types, including colon, lung, head and neck, ovarian, and renal cell carcinomas.[2]
Two monoclonal antibodies (MoAbs) that target EGFR are currently available for clinical use, panitumumab and cetuximab. These MoAbs competitively inhibit the binding of EGFR by natural ligands.[3] Panitumumab is currently indicated for the treatment of patients with EGFR-expressing metastatic colorectal carcinoma (mCRC) with disease progression on or after fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens.[4] Cetuximab, a chimeric MoAb, is indicated as a single agent and in combination with irinotecan for the treatment of patients with EGFR-expressing refractory mCRC who have failed or are intolerant of irinotecan and as a single agent or with radiation therapy in patients with squamous cell head and neck cancers.[5]
Panitumumab is the first recombinant, fully human MoAb that binds to EGFR on both normal cells and tumor cells.[6,7] In preclinical studies, panitumumab has been shown to block various EGFR functions.[7-9] Fully human antibodies have the potential to result in lower immunogenicity compared with chimeric antibodies.[10]
The objectives of this study were to evaluate the safety, pharmacokinetics, and efficacy of 2 dose schedules and 2 infusion durations of panitumumab in patients with solid tumors. In previous studies, panitumumab infusions were administered over 60 minutes with a low incidence of infusion reactions. Because a reduced duration of panitumumab administration (ie, over 30 minutes instead of 60 minutes) has the potential to be more convenient for the patient and decrease caregiver time, the 30-minute infusion time was evaluated in this study.
Patients and Methods
Patient Eligibility
Patients were eligible if they were aged ≥ 18 years, capable of providing informed consent, and had a pathologic diagnosis of an advanced solid tumor that was refractory to ≥ 1 standard therapies or for which no standard therapy exists. Patients could not have received > 3 previous treatment regimens (excluding hormonal therapies for breast and prostate cancers). They had to have measurable or nonmeasurable disease per Response Evaluation Criteria in Solid Tumors (RECIST);[11] Eastern Cooperative Oncology Group (ECOG) status of 0 or 1; life expectancy of ≥ 3 months; tumor tissue available for immunohistochemistry studies for EGFR expression; and adequate hematologic, renal, and hepatic functions. Patients who had a history of other primary cancers were eligible only if they had the following: nonmelanoma skin cancers not requiring treatment or had curative resection, curatively treated cervical carcinoma in situ or other primary solid tumors, no treatment administered in the previous 3 years, and no active disease within the previous 5 years. Previous treatment with an anti-EGFR antibody was not permitted.
Study Design
This phase I multicenter, open-label study evaluated panitumumab administered alone in patients with advanced solid malignancies refractory to standard therapy or for which no standard therapy exists. This study compared 2 dosages and schedules of panitumumab: 6 mg/kg infused every 2 weeks and 9 mg/kg administered every 3 weeks. Two infusion schedules were evaluated in patients receiving 6 mg/kg every 2 weeks: all infusions for 60 minutes (cohort 1A) or the first infusion of panitumumab over 60 minutes and subsequent infusions delivered over 30 minutes (cohort 1B) if the first infusion was well tolerated (ie, no serious infusion reactions were observed). Patients were enrolled sequentially into cohort 1A, then into cohort 1B. Enrollment for cohorts 1A and 1B was completed before beginning enrollment in cohort 2, which included patients receiving panitumumab 9 mg/kg every 3 weeks with all infusions delivered over 60 minutes. An interim analysis of safety was performed after the first 10 patients in cohort 1A had completed study week 8. Patients continued panitumumab therapy until disease progression, inability to tolerate therapy, or withdrawal from the study for any other reason. A safety follow-up was scheduled 4 weeks after the last dose of panitumumab.
The study was conducted from August 2004 through April 2006. This trial was registered under the US National Institutes of Health ClinicalTrials.gov identifier NCT00091806. This study was conducted in compliance with the Declaration of Helsinki. The study protocol and informed consent forms were approved by all institutional review boards, and all patients signed a written consent form before initiation of study-specific procedures.
Endpoints and Asessments
The primary safety endpoint was the incidence of grade 3 and 4 adverse events, which were graded according to the National Cancer Institute Common Toxicity Criteria, version 2.0, with the exception of prespecified skin/dermatologic toxicities, which were graded using Common Terminology Criteria for Adverse Events, version 3.0, with modifications. Additional primary safety endpoints were the incidence of serious adverse events and events leading to discontinuation of panitumumab. Secondary safety endpoints included the incidence of antipanitumumab antibody formation and incidence of adverse events of any grade.
A post hoc analysis was performed to identify adverse events that might have indicated a hypersensitivity reaction to panitumumab. These events included preferred terms for allergic reaction/hypersensitivity and cytokine release syndrome/acute infusion reaction categories, and the events had to be temporally related to the panitumumab infusion.
Serum samples to be tested for the presence of antipanitumumab antibodies were collected within 30 minutes before the panitumumab infusion at weeks 1, 3, 5, 7, and every 8 weeks thereafter for patients in cohort 1 and at weeks 1, 4, 7, 10, and every 9 weeks thereafter for patients in cohort 2. Antipanitumumab antibodies were measured using 3 validated assays. Serum samples were screened with a bridging enzyme-linked immunosorbent assay (ELISA) that included an acid dissociation sample preparation step designed to reduce interference from excess panitumumab in the patient samples. A BiacoreTM (Biacore International, AB, Uppsala, Sweden) assay was used as the second screening assay. Any samples that tested positive for antipanitumumab antibodies in either of the screening assays were further tested for neutralizing antibodies in a cell-based EGFR phosphorylation assay. All antipanitumumab antibody assays were performed by Amgen Inc (Thousand Oaks, CA).
An additional primary objective of the study was to assess the pharmacokinetics of panitumumab at these dose schedules. Serum samples for pharmacokinetic analyses were collected within 15 minutes before and 30 minutes after completion of the panitumumab infusion in weeks 1, 3, 5, and 7, and every 8 weeks thereafter for patients in cohort 1 and in weeks 1, 4, 7, and 10, and every 9 weeks thereafter for patients in cohort 2. In addition, serum samples were collected from approximately 20 patients from each cohort at 24, 96, and 168 hours, and 240 hours (cohort 1) and 336 hours (cohort 2) after the first and third infusions. A validated immunoassay with electrochemiluminescence detection was used to measure panitumumab concentrations and was performed by Amgen Inc (Fremont, CA). Standard noncompartmental pharmacokinetic analyses were performed using WinNonlin Professional, version 4.1e (Pharsight Corp, Mountain View, CA). The pharmacokinetic endpoints included the minimum (Cmin) and maximum (Cmax) observed serum concentrations of panitumumab, area under the serum concentration-time curve (AUC), panitumumab half-life (t1/2) within the dosing interval, and serum clearance after intravenous (I.V.) administration.
Efficacy endpoints included the incidence of objective responses (complete or partial responses, stable disease, or progressive disease) and duration of responses. Tumor responses were assessed per RECIST guidelines[11] at weeks 8, 16, 24, 32, 40, and 48. Patients who discontinued without a postbaseline tumor assessment or with an unconfirmed complete response or partial response were considered to be nonresponders.
Statistical Analyses
The planned sample size was 85 patients (20 patients in cohort 1A; 45 patients in cohort 1B; and 20 patients in cohort 2), which was calculated based on the probability of observing. 1 of the adverse events listed in the early study stopping guidelines (assuming an underlying rate of 5%). Adverse events listed in the early study-stopping guidelines included certain skin toxicities (symptomatic skin-related toxicity requiring narcotics or systemic steroids or considered intolerable by the patient; skin infection requiring systemic I.V. antibiotic or I.V. antifungal treatment; need for surgical debridement; or any skin-related serious adverse event); grade 3/4 diarrhea; grade 3/ 4 cardiac events; or any other serious adverse events determined to be related to panitumumab.
An exploratory analysis was performed to compare pharmacokinetic profiles of patients receiving panitumumab produced on a 2-kL scale (historical data) with patients receiving 12-kL-scale product in this study. Log-transformed AUC0-tau and Cmax values after the first and third doses were analyzed using a one-factor analysis of variance (equivalent to a t test). The mean difference and 90% CI for the mean difference were also determined. These statistical analyses were performed using SAS, version 8.2 (SAS Institute Inc, Cary, NC).
Efficacy analyses were conducted for all enrolled patients who had measurable disease at baseline and received panitumumab. For discrete data, the frequency and percentage distributions were provided. For continuous endpoints, the mean, standard deviation, median, minimum, and maximum values were provided. For patients with an objective response, the duration of response (time from initial documentation of the response to disease progression or death) was summarized by a Kaplan-Meier curve and 95% confidence intervals (CIs) were calculated. The final analyses were performed when all patients had completed end-of-study assessments.
Study Drug
Panitumumab (supplied by Amgen Inc, Thousand Oaks, CA) used in this study was produced from Chinese hamster ovary (CHO) cells on a 12-kL scale, in contrast with previous studies that used panitumumab manufactured on a 2-kL scale.[12-14] Panitumumab was supplied in 10-mL vials at a concentration of 20 mg/mL. The appropriate volume of panitumumab was diluted in 100 mL of normal saline and administered using a 0.22-µ in-line filter.
Patient Disposition and Demographics
An interim safety analysis was performed after the first 10 patients enrolled in the study had completed study week 8. Too few events listed in the early study-stopping guidelines were noted in this interim analysis, and enrollment in the study was allowed to proceed. All cohorts were fully enrolled as planned. The disposition of all patients who were screened is detailed in Table 1 .
From 8 centers in the United States, 86 patients were enrolled; 84 of the patients (98%) received ≥ 1 dose of panitumumab and were included in the safety analysis set. Two patients did not receive panitumumab because of investigator decision or withdrawn consent. The most common reason for discontinuation was disease progression (as evidenced by radiographic documentation). Reasons for discontinuation from the study included disease progression (57%), adverse event (6%), patient request (6%), administrative decision (6%), death (5%), protocol-specified criterion (1%), determination of ineligibility (1%), and other (16%). Most patients (63%) completed a safety follow-up 4 weeks after the last infusion of panitumumab; reasons for not completing the safety follow-up included death (15%), disease progression (8%), adverse event (2%), withdrawn consent (2%), loss to follow-up (1%), and other (8%). The mean duration of follow-up for all patients was 13.2 weeks (standard deviation [SD], 11 weeks).
The patients were predominantly male and white, and the mean age was 60 years (range, 21-83 years; Table 2 ). The most common tumor types were non–small-cell lung, colorectal, and esophageal cancers. Tumor types were not equally represented in both cohorts. A higher proportion of patients in cohort 2 had an ECOG score of 1 than in the other cohorts. Six patients with an ECOG status of 2 were enrolled in the study before an amendment to the protocol that required patients to have an ECOG status of 0 or 1.
Panitumumab Exposure
Patients in cohort 1 received more infusions than patients in cohort 2. The mean number of infusions received was 5.8 (SD, 4.5 infusions) for patients in cohort 1A, 5.2 (SD, 4.6 infusions) for patients in cohort 1B, and 5.4 (SD, 4.6 infusions) for all patients in cohort 1. Patients in cohort 2 had a mean number of 2.8 infusions (SD, 2.1 infusions). The cumulative exposure to panitumumab was correspondingly higher in patients receiving panitumumab every 2 weeks. The mean cumulative exposure was 34 mg/kg, 31 mg/kg, and 24 mg/kg in patients in cohorts 1A, 1B, and 2, respectively.
Safety
All dosed patients had ≥ 1 adverse event during the study ( Table 3 ). Most events were grades 1 to 3 in severity. Twenty-three patients (27%) discontinued the study because of an adverse event; however, only 5 patients (6%) discontinued the study because of a treatment-related adverse event as determined by the investigator.
Most patients (90%) in all cohorts had ≥ 1 adverse event related to the treatment with panitumumab ( Table 3 ). Most patients had grade 1 (21%) or grade 2 (40%) events. Treatment-related adverse events ( Table 4 ) were generally consistent with the known effects of EGFR inhibitors, including skin-related adverse events (erythema, pruritus, acneiform dermatitis, and rash) and fatigue. Skin-related events were all grades 1 to 3 in severity across all cohorts. The incidence of grade 3 events was somewhat higher in patients in cohort 2 (62%) than in patients in cohort 1 (37%). For adverse events occurring in > 10% of patients, grade ≤ 3 adverse events of erythema (3% in cohort 1 vs. 10% in cohort 2), pruritus (3% vs. 10%), acneiform dermatitis (6% vs. 19%), fatigue (2% vs. 14%), and hypomagnesemia (5% vs. 10%) were less frequent in patients in cohort 1 than in cohort 2. However, the incidence of adverse events of any grade was commonly higher in cohort 1 than in cohort 2, including erythema, pruritus, acneiform dermatitis, nail disorder/paronychia, nausea/vomiting, anorexia, dry skin, and diarrhea. There were no grade 4 or 5 treatment-related adverse events.
There were no investigator-reported infusion reactions; however, in a conservative post hoc analysis, 5 potential infusion reactions occurred in 4 patients (5%). These potential infusion reactions occurred in 3 patients (7%) in cohort 1B (all of whom had metastatic non–smallcell lung cancer) and 1 patient (5%) in cohort 2. Overall, 1% (5/398) of all infusions (including both 30- and 60-minute infusions) were associated with potential infusion reactions, and 2% (4 of 181) of the 30-minute infusions were associated with a potential infusion reaction. All 5 of the potential infusion reactions occurred at the second or later infusions; none of the infusions were stopped because of the potential infusion reaction. One patient discontinued panitumumab following the potential infusion reaction because of disease progression. The other 3 patients had additional infusions that were well tolerated. Two of the patients did not receive premedication for subsequent infusions.
Immunogenicity
Baseline (predose) antibody samples were collected from 83 patients (99%) who received panitumumab and ≥ 1 postdose samples were collected from 76 patients. Using the Biacore assay, postdose antipanitumumab antibodies were detected in 5 patients (6.6%). Two of these 5 patients (2.6%) also tested positive for neutralizing antibodies. Pre-existing (ie, predose) antibodies were detected in 3 of the 5 patients who tested positive for antipanitumumab antibodies in the Biacore assay. By ELISA, only 1 patient tested positive for postdose (transiently, only at week 5) antipanitumumab antibodies; this patient tested positive in the Biacore assay and the bioassay for neutralizing antibodies.
Pharmacokinetics
Serum panitumumab concentration-time profiles after the first and third doses of panitumumab are shown in Figure 1. After the third dose, when panitumumab pharmacokinetics were at steady state, the exposure to panitumumab (AUC) increased in an approximately dose-proportional manner: the mean was 1310 µg day/mL (SD, 375 µg day/mL) for patients in cohort 1 and 1790 µg day/mL (SD, 794 µg day/mL) for patients in cohort 2 ( Table 5 ). The median peak concentration of panitumumab for patients receiving 30-minute infusions (180 µg/mL) was similar to that for patients receiving 60-minute infusions (177 µg/mL; Figure 2). 
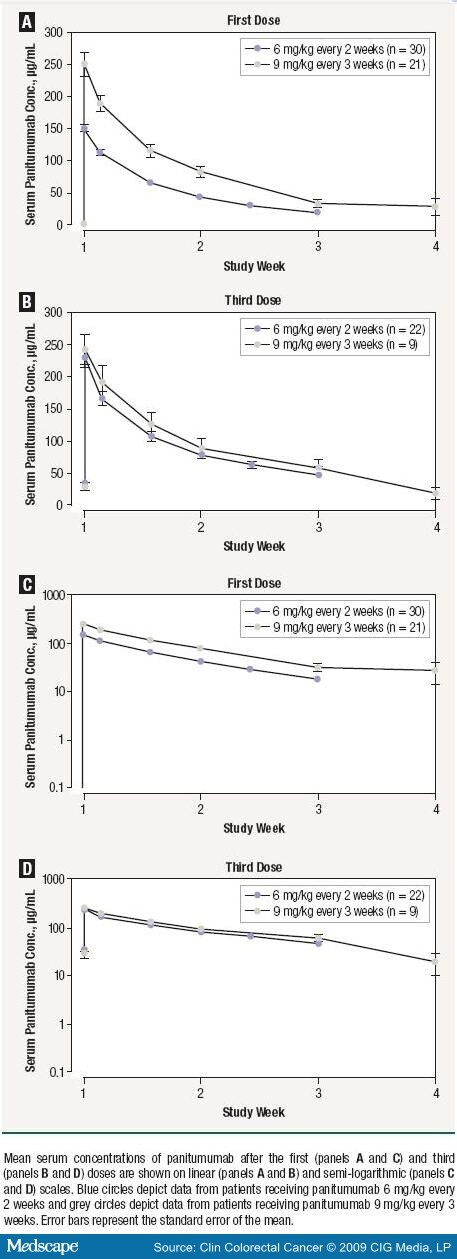
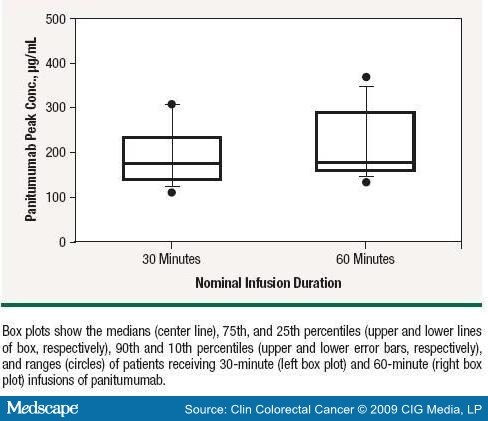
Efficacy
The efficacy analysis set included 82 (95%) patients who had measurable disease at baseline. Four patients (5%) had an objective response while on study: 1 patient in cohort 1A and 3 patients in cohort 1B. All were partial responses. Responses were seen in the following tumor types (1 patient each): colon cancer (cohort 1A), bladder cancer, esophageal cancer, and rectal cancer (cohort 1B). The median duration of response was 19.1 weeks (95% CI, 12.6-25.6 weeks) for responders in cohort 1B. For the single patient in cohort 1A who had an objective response, the duration of response was 32.3 weeks. Thus, the overall median (minimum, maximum) duration of response for all patients was 25.6 weeks.13,26 There were no objective responses in the patients in cohort 2. Twenty-three patients had stable disease of ≥ 8 weeks, including 17 patients in cohort 1 and 6 patients in cohort 2.
Discussion
This study was designed to evaluate the safety, pharmacokinetics, and efficacy of 2 dosages and 2 infusion times of panitumumab in patients with solid tumors. The overall safety profile of panitumumab at doses of 6 mg/kg every 2 weeks and 9 mg/kg every 3 weeks was generally consistent with that observed in other clinical trials.[6,7,12,13,15] The adverse event profile was similar when panitumumab was administered at doses of 6 mg/kg every 2 weeks (cohort 1) or 9 mg/kg every 3 weeks (cohort 2), although the severity of adverse events was greater among patients in patients in cohort 2. Skin-related adverse events were most common, comprising 4 of the 5 most commonly reported adverse events. Skin-related events were mostly grades 1 to 3 in severity, and no life-threatening or fatal skin-related adverse events were reported. The overall safety profile of 12-kL CHO-derived panitumumab at these dosing regimens was consistent with that observed in previous clinical studies of panitumumab evaluating the 2-kL material, as measured by the incidence of related adverse events.
Differences in drug exposure were noted in patients receiving panitumumab every 2 weeks and every 3 weeks. Notably, patients receiving every-2-week infusions had a higher exposure to panitumumab than patients receiving every-3-week infusions; the difference in drug exposure might have been because the frequency of dosing resulted in delivery of a larger number of infusions to patients in cohort 1 (every 2 weeks) over the course of the study.
Treatment-related adverse events were generally consistent with the known effects of EGFR inhibitors, including skin-related adverse events and fatigue. The incidence of some treatment-related adverse events (anorexia, diarrhea), including some skin-related adverse events (erythema, pruritus, dry skin), was higher in patients receiving panitumumab every 2 weeks, which might reflect the frequency of dosing in patients in cohort 1. Grade 3 events of erythema, pruritus, acneiform dermatitis, fatigue, and hypomagnesemia were higher in patients in cohort 2, possibly because of the higher dose-proportional drug exposure. There also was no obvious difference in the grade 4 or 5 adverse events between cohorts 1 and 2, although an intercohort comparison of safety is limited by the sample size. There were no grade 4/5 treatment-related adverse events.
In this study, no differences in safety were noted when panitumumab 6 mg/kg every 2 weeks was administered as a 60-minute infusion or as a 60-minute infusion followed by 30-minute infusions. Both the 30-minute and 60-minute infusions were well tolerated, with an incidence of potential infusion reactions (2% of 30-minute infusions and 1% of 60-minute infusions) similar to that observed in other clinical trials of panitumumab.[4] A reduced duration of panitumumab administration might provide greater convenience for both patients and caregivers.
Many therapeutic proteins are associated with an increased risk of hypersensitivity reactions, especially MoAbs that are chimeric or humanized, even when the murine portion of a chimeric or humanized MoAb is proportionally small.[10] Traditionally, initial infusion times have been lengthy to minimize drug exposure while monitoring the patient for an infusion reaction. Cetuximab is a chimeric construct, is administered as an initial 120-minute infusion, and require premedication with an antihistamine.[5] Serious infusion reactions have been reported in 2%-5% of patients receiving cetuximab in clinical trials,[5] and rates of grade 3/4 infusion reactions with cetuximab have been reported to be as high as 22% in some patient populations in the southeastern United States.[16]
Grade 3/4 infusion reactions have occurred in approximately 1% of patients receiving panitumumab in clinical trials.[4] Although no infusion reactions were reported by the investigators during the course of this study, a post hoc analysis was performed to identify adverse events that may have indicated a hypersensitivity reaction to panitumumab. It should be noted that some signs and symptoms used in this analysis are commonly seen in patients with cancer. This analysis revealed 5 potential infusion reactions in 4 patients. All of the potential infusion reactions occurred at the second infusion or later infusions, in contrast with typical infusion reactions to cetuximab, which mostly occur (90%) with the first infusion, despite premedication with antihistamines.[5]
Antipanitumumab antibodies were detected in 6.6% of patients with available postdose samples; 2.6% tested positive for neutralizing antibodies. The incidence of binding antibodies to panitumumab in this trial is similar to rates seen in previous clinical trials of panitumumab. 4 Although the number of patients with antipanitumumab antibodies was small, the safety data did not reveal any trends indicative of an altered panitumumab safety profile for patients with neutralizing or non-neutralizing antipanitumumab antibodies.
An approximate dose-proportional increase in exposure to panitumumab was observed after the third dose, when panitumumab pharmacokinetics were at a steady state. No apparent differences in the peak serum concentrations of panitumumab were observed between the 30- or 60-minute infusions. The peak concentration is not sensitive to infusion duration as the infusion process for panitumumab is significantly faster than the distribution and elimination phases (half-lives in days).
Panitumumab pharmacokinetics from this study were similar to the results from a phase 1 study using drug manufactured on a 2-kL scale.12 In this study, which evaluated panitumumab produced at a 12-kL scale, pharmacokinetic profiles after the first and third doses of 6 mg/kg every-2-weeks panitumumab were similar to those for panitumumab produced at a 2-kL scale. Although these studies were not designed or powered as bioequivalence studies, the 90% CIs of the ratio of the geometric means for Cmax and AUC0-tau were within or slightly outside the 80% to 125% interval used as a criterion of bioequivalence by the US Food and Drug Administration (FDA). The 90% CI for the AUC ratio after the third dose (83% to 121%) was within the interval (80% to 125%). Although the 90% CI for the Cmax ratio after the third dose (83% to 130%) extended slightly beyond the 125% limit, the point estimate was favorable (108%). These small differences are not likely to be clinically significant.
Antitumor activity with the 6-mg/kg every-2-week dose schedule of panitumumab was observed. Objective responses to panitumumab were seen in 4 patients (5%); this rate is consistent with rates observed in other clinical trials of panitumumab alone (5% to 10%).13,14,17 The tumor types seen in patients in cohort 2 were different from the tumor types seen in cohort 1, which might account for the different response rates in the 2 cohorts. Notably, no patients with colorectal cancers were enrolled in cohort 2. Panitumumab has demonstrated efficacy and is FDA approved for the treatment of patients with mCRC.4,14 In our study, half of the patients with an objective response had colon or rectal cancer. The remaining objective responses were seen in patients with bladder and esophageal cancers, supporting the need for further evaluation of panitumumab in the treatment of patients with other solid tumors. The activity of the 12-kL material appeared to be consistent with that seen with the 2-kL preparation used in previous studies.
